|



| Research |
| | | |
 If you are interested in working with us on the GIT microbiome, please contact: Andrés Moya Amparo Latorre | The GIT microbiome in health & disease The knowledge about bacterial diversity in the human GIT has increased concomitantly with the development of different molecular approaches such as cloning and subsequent sequencing of the 16S rRNA genes, fingerprinting techniques of 16S rDNA amplicons (DGGE, TTGE, etc.), fluorescent in situ hybridization, DNA microarray or recently massive sequencing. The composition of the resident intestinal microbiota varies between individuals due to host genotype, age, health status and diet, with the predominant populations being fairly stable under normal conditions. Furthermore, the human gut microbiome presents many physiological properties that are lacking in the host and thus can be considered as essential for human life. Thus, it is necessary to understand the functional significance of the gut microbiota in human health and well-being as well as the microbe-microbe or microbe-host interactions. We apply a multidisciplinary approach to characterize, using metagenomic and metatranscriptomic tools, the normal microbiota of the human gut in human populations to understand the conditions that facilitate or prevent colonization by exogenous microorganisms, to study its evolution during acute stress (antibiotic treatment), to explore changes in diseases affecting the gut ecology, and to establish reliable methods for its restoration. This is expected to significantly improve our knowledge of the human-microbiota relationships in health and disease, proposing directed interventions to restore these relationships for situations in which the equilibrium is disrupted. | |
| | | |
 If you are interested in working with us on developmental metagenomics of the GIT microbiome, please contact: M. Pilar Francino | Development of the GIT microbiome in infants During the course of the first year of life, the infant´s GIT progresses from harboring a low number and diversity of microbial inhabitants to sustaining an extremely dense microbial community, ending with a mixture of microbes that approaches the one found in the adult´s GIT. This process is far from a strict microbial succession; rather, GIT colonization is highly variable among individuals and is influenced by numerous factors. Our work studies the process of development of the GIT microbiome during infancy, by analyzing the taxonomic composition, coding capabilities and gene expression patterns of this community at different stages, from birth to the end of the infant´s first year. We are also interested in the relationship between GIT microbiome development and health. With this perspective, we are investigating the repertoire of antibiotic resistances and related phenotypes in the infant´s GIT, the interactions between microbiome and immune system development, and the potential associations that some of these interactions may have with the onset of atopic diseases. | |
| | | |
 If you are interested in working with us on the GIT viral metagenome, please contact: Andrés Moya | The GIT viral metagenome The human gastrointestinal tract is the natural habitat for a large microbial community including species from the kingdoms Archaea, Bacteria, and Eukarya. The gastrointestinal microbiota also contains enteric viruses, including a variety of bacteriophages and a number of known human viruses and of uncharacterized viruses. Identifying and measuring the community dynamics of viruses in the environment is complicated because less than one percent of microbial hosts have been cultivated. Also, there is no single gene that is common to all viral genomes, so total uncultured viral diversity cannot be monitored using approaches analogous to ribosomal DNA profiling commonly used for Bacteria and Archaea. Alternative approaches are therefore required for the evaluation of viral consortia in environmental samples. While traditional sequencing approaches are designed to characterize genomes of a single or few species of interest, metagenomic approaches of uncultured viral communities, such as mass sequencing, allow us to explore the makeup of microbial communities. Such methods provide a holistic look at microbial diversity within a given sample, completely bypassing the need for culturing. So far, most of the metagenomic studies on viriomes have been focused on non-human environments, such as fresh or salt water ecosystems. Those dealing with human viriomes have been partial and specifically interested in subsets of viruses, on small numbers of individuals, mostly healthy ones. Metagenomic studies on human viruses related to disease have been rare and restricted to acute | |
| | | |
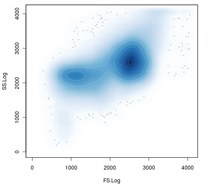 If you are interested in working with us onFlow cytometry and single-cell genomics, please contact: Giuseppe D´Auria | Flow cytometry and single-cell genomics Flow cytometry cell sorting coupled with fluorescent hybridization techniques enables the separation of microbial populations of interest from complex samples independently of cultivation. In the last years this technology has been successfully employed to describe subpopulations from complex environmental samples as well as for advanced single-cell genomic studies. The research line in "Flow cytometry and single-cell genomics" aims to develop and apply protocols for microbial cell sorting and single-cell genomics. The main objectives of the research line include: 1) To obtain clear taxonomical descriptions of complex microbial communities with special attention to underrepresented microbial fractions. 2) To study the microbial diversity in patients affected by gastrointestinal disease such as Clostridium difficile-associated colitis, inflammatory bowel disease, Crohn, etc. 3) To identify recurrent players or disease-associated organisms. 4) To promote single-cell genomics studies based on flow cytometry enrichments when cultivation of the organism is not possible. 5) To carry out metagenomics and metatranscriptomics analysis based on population enrichments obtained by fluorescent activated cell sorting. The research line also promotes the development of new bioinformatics analytical tools needed to cope with the high number of sequences provided by next generation sequencing technologies. | |
| | | |